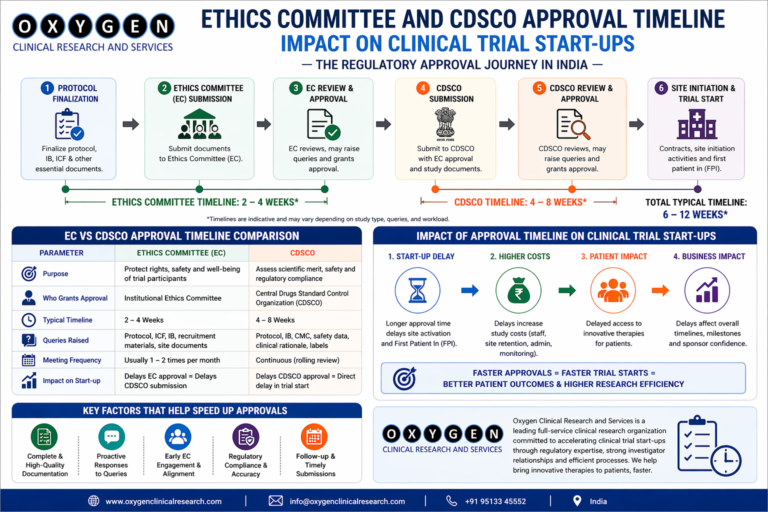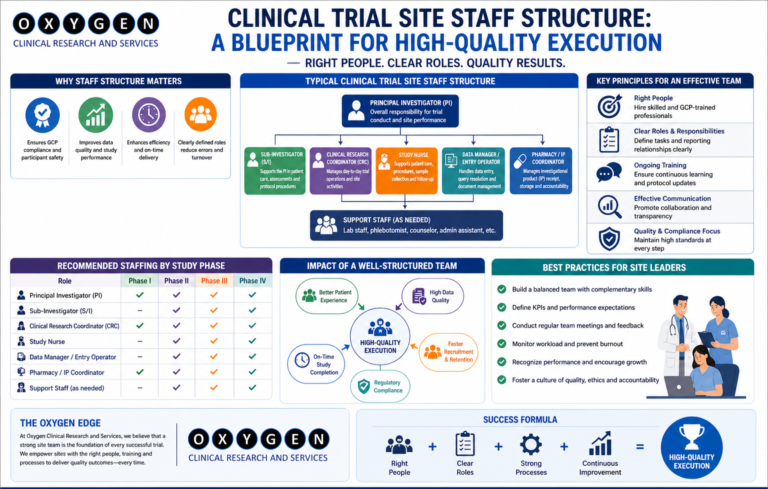
Clinical Trial Site Staff Structure: A Blueprint for High-Quality Execution
Clinical Trial Staff Structure. Most clinical trial delays in India do not stem from a lack of patients; they stem from a lack of bandwidth at the site level. When I review failed timelines, the common denominator is almost always an under-resourced site team, where the Principal Investigator (PI) is distracted by clinical practice and the study coordinator is overwhelmed by administrative burden. If you are a sponsor or a CRO leader, you know the cost of a site falling behind. It is not just the burn rate of the site; it is the delay in your global submission, the integrity of your data, and the potential for a disastrous audit. After 15 years in the field, I have realized that the site staff structure is the most critical variable in the trial success equation. The following comparison highlights the impact of site staffing models on key trial metrics. Sr No Role Structure Budget Impact Start-up Speed Data Quality Audit Risk Protocol Adherence Staff Turnover Patient Retention Monitoring Load 1 Full-time Dedicated High Fast High Low Excellent Low High Medium 2 Part-time Shared Medium Moderate Medium Medium Good Moderate Medium High 3 Minimalist/PI-led Low Slow Low High Poor High Low Very High 4 Hybrid (CRO-funded) High Fast High Low Excellent Low High Medium 5 Outsourced Support Medium Moderate Medium Medium Moderate High Moderate High The Reality of Site Staffing in India Under the New Drugs and Clinical Trials Rules (2019), the responsibility of the PI is absolute. However, the operational execution is delegated. A standard, high-performing site team in India requires more than just a PI and a coordinator. Essential Roles for Execution When these roles are collapsed—for example, when a single person handles both IP management and patient recruitment—data quality suffers. We see this often in clinical trial site management in India. Operational Bottlenecks: Where Projects Fail Delays occur at the intersection of regulatory compliance and site workload. CDSCO and EC submissions are rigorous. If your site team is focused only on patient visits, the regulatory paperwork—the foundation of your trial—will lag. Table 2: Common Operational Failures at Site Level Sr No Process Area Typical Delay Impact on Trial Root Cause Risk Level Mitigation Strategy Cost Overrun 1 EC Approval 4–6 weeks Start-up Incomplete docs High Proactive review Low 2 IP Management Ongoing Compliance Poor documentation Critical Dedicated staff Moderate 3 Data Entry 2+ weeks Query load Staff bandwidth Medium Real-time monitoring High 4 Patient Consent Daily Ethics Hurried process Critical Training/Audit High 5 Lab/Sample Ongoing Data loss Improper logs Critical Standardized SOPs High Case Studies: Real-World Execution Insights Case 1: The Overloaded Coordinator Case 2: The Regulatory Bottleneck Case 3: The IP Compliance Breach Challenges and Mitigation: The Hard Truth Most sponsors try to save costs by minimizing site staff. This is a false economy. A site with a lean, overworked team will require more monitoring visits (CRAs), more data cleaning, and potentially more site management travel. My experience at Oxygen Clinical Trial shows that investing in the right headcount at the start prevents expensive “firefighting” later. Myths vs. Reality Practical Sponsor Checklist Feasibility Stage Startup Stage Execution Stage Regulatory Context: CDSCO and Global Standards Compliance with the New Drugs and Clinical Trials Rules (2019) is non-negotiable. Your site staff must be trained on ICH-GCP E6(R3) and understand the implications of non-compliance for both the sponsor and the investigator. Periodic review of CTRI registration and adherence to EC conditions is where many projects fail during inspections. FAQ: Clinical Operations Insights Why does site monitoring often fail at the start? Lack of alignment on expectations. The CRA expects the site to be ready, but the site lacks the administrative support to prioritize the CRA’s requests Clinical Trial Staff Structure. Conclusion Effective site staff structure is the difference between a high-quality data set and a regulatory warning letter. If you are struggling with site performance or want to ensure your next study in India is executed with precision, let’s discuss your operational strategy. For site management support, you can reach out directly at govindpawar@oxygenclinicaltrials.com or connect with me on LinkedIn. You can also learn more about our approach at Oxygen Clinical Trial. For specific inquiries, use our contact page.